Parsing alignments into pairs¶
Overview¶
Hi-C experiments aim to measure the frequencies of contacts between all pairs of loci in the genome. In these experiments, the spacial structure of chromosomes if first fixed with formaldehyde crosslinks, after which DNA is partially digested with restriction enzymes and then re-ligated back. Then, DNA is shredded into smaller pieces, released from nucleus, sequenced and aligned to the reference genome. The resulting sequence alignments reveal if DNA molecules were formed through ligations between DNA from different locations in the genome. These ligation events imply that ligated loci were close to each other when the ligation enzyme was active, i.e. they formed “a contact”.
pairsamtools parse detects ligation events in the aligned sequences of
DNA molecules formed in Hi-C experiments and reports them in the .pairs/.pairsam
format.
Terminology¶
Throughout this document we will be using the same visual language to describe how DNA sequences (in the .fastq format) are transformed into sequence alignments (.sam/.bam) and into ligation events (.pairs).

DNA sequences (reads) are aligned to the reference genome and converted into ligation events¶
Short-read sequencing determines the sequences of the both ends (or, sides) of DNA molecules (typically 50-300 bp), producing read pairs in .fastq format (shown in the first row on the figure above). In such reads, base pairs are reported from the tips inwards, which is also defined as the 5’->3’ direction (in accordance of the 5’->3’ direction of the DNA strand that sequence of the corresponding side of the read).
Alignment software maps both reads of a pair to the reference genome, producing alignments, i.e. segments of the reference genome with matching sequences. Typically, there will be only two alignments per read pair, one on each side. But, sometimes, the parts of one or both sides may map to different locations on the genome, producing more than two alignments per DNA molecule (see Multiple ligations (walks)).
pairsamtools parse converts alignments into ligation events (aka
Hi-C pairs aka pairs). In the simplest case, when each side has only one
unique alignment (i.e. the whole side maps to a single unique segment of the
genome), for each side, we report the chromosome, the genomic position of the
outer-most (5’) aligned base pair and the strand of the reference genome that
the read aligns to. pairsamtools parse assigns to such pairs the type UU
(unique-unique).
Unmapped/multimapped reads¶
Sometimes one side or both sides of a read pair may not align to the reference genome:

A read pair missing an alignment on one or both sides¶
In this case, pairsamtools parse fills in the chromosome of the corresponding
side of Hi-C pair with !, the position with 0 and the strand with -.
Such pairs are reported as type NU (null-unique, when the other side has
a unique alignment) or NN (null-null, when both sides lack any alignment).
Similarly, when one or both sides map to many genome locations equally well (i.e.
have non-unique, or, multi-mapping alignments), pairsamtools parse reports
the corresponding sides as (chromosome= !, position= 0, strand= -) and
type MU (multi-unique) or MM (multi-multi) or NM (null-multi),
depending on the type of the alignment on the other side.

A read pair with a non-unique (multi-) alignment on one side¶
pairsamtools parse calls an alignment to be multi-mapping when its
MAPQ score
(which depends on the scoring gap between the two best candidate alignments for a segment)
is equal or greater than the value specied with the --min-mapq flag (by default, 1).
Multiple ligations (walks)¶
Finally, a read pair may contain more than two alignments:
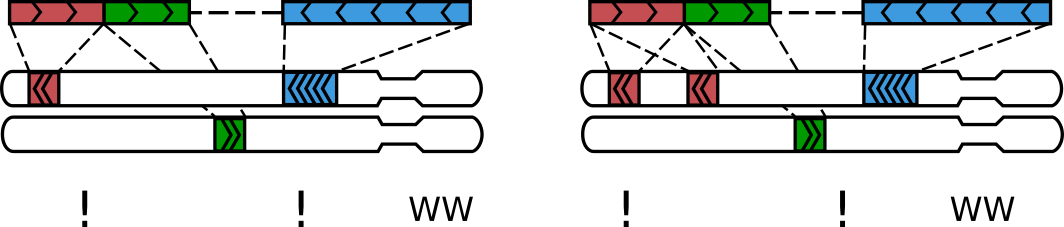
A sequenced Hi-C molecule that was formed via multiple ligations¶
Molecules like these typically form via multiple ligation events and we call them
walks 1. Currently, pairsamtools parse does not
process such molecules and tags them with type WW. Note that, each of the
alignments
Interpreting gaps between alignments¶
Reads that are only partially aligned to the genome can be interpreted it in two different ways. One possibility is to assume that this molecule was formed via at least two ligations (i.e. it’s a walk) but the non-aligned part (a gap) was missing from the reference genome for one reason or another. Another possibility is to simply ignore this gap (for example, because it could be an insertion or a technical artifact), thus assuming that our molecule was formed via a single ligation and has to be reported:
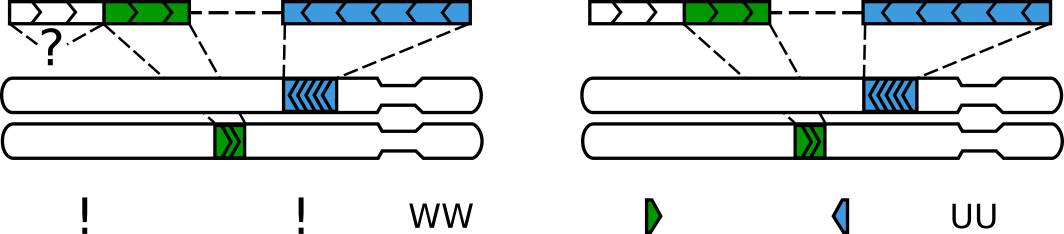
A gap between alignments can interpeted as a legitimate segment without an alignment or simply ignored¶
Both options have their merits, depending on a dataset, quality of the reference
genome and sequencing. pairsamtools parse ignores shorter gaps and keeps
longer ones as “null” alignments. The maximal size of ignored gaps is set by
the --max-inter-align-gap flag and, by default, equals 20bp.
Rescuing single ligations¶
Importantly, some of DNA molecules containing only one ligation junction may still end up with three alignments:
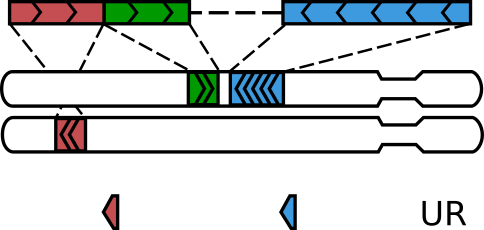
Not all read pairs with three alignments come from “walks”¶
A molecule formed via a single ligation gets three alignments when one of the two ligated DNA pieces is shorter than the read length, such that that read on the corresponding side sequences through the ligation site and into the other piece 2. The fraction of such molecules depends on the type of the restriction enzyme, the typical size of DNA molecules in the Hi-C library and the read length, and sometimes can be considerable.
pairsamtools parse detects such molecules and rescues them (i.e.
changes their type from a walk to a single-ligation molecule). It tests
walks with three aligments using three criteria:
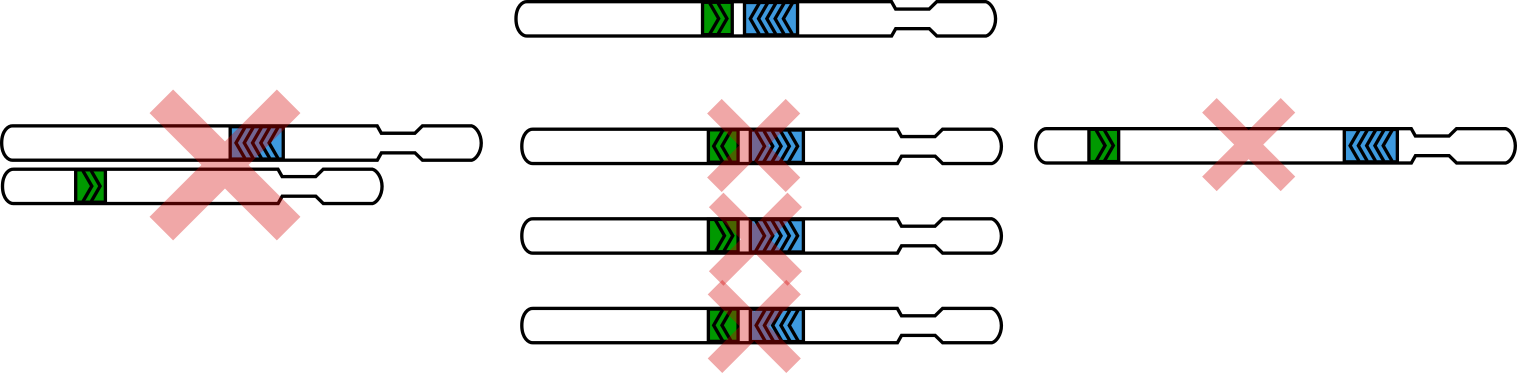
The three criteria used for “rescue”¶
On the side with two alignments, the “inner” one must be on the same chromosome as the alignment on the other side.
The “inner” alignment and the alignment on the other side must point toward each other.
These two alignments must be within the distance specified with the
--max-molecule-sizeflag (by default, 2000bp).
Sometimes, the “inner” alignment is non-unique or “null” (i.e. when the unmapped
segment is longer than --max-inter-align-gap, as described in Interpreting gaps between alignments).
pairsamtools parse rescues such walks as well.
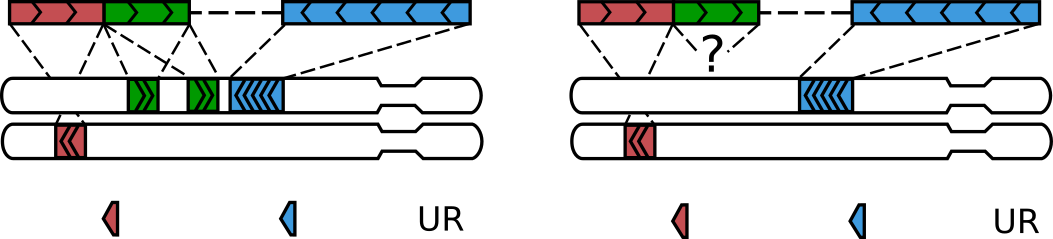
A walk with three alignments get rescued, when the middle alignment is multi- or null.¶